Inferências filogenéticas são procedimentos de estimativas. Assim, existe uma série de algoritmos que podem ser aplicados em estimativas filogenéticas e que são classificados em duas categorias:
A) critérios quantitativos: baseados em distância entre os dados:
- distância média [UPGMA - "unweighted pair group method with arithmetic means"] (Sokal & Michener, 1958; Sneath & Sokal, 1973);
- distância transformada [DT] (Farris, 1977);
- método de Fitch e Margoliash (1967);
- evolução mínima [EM] (Cavalli-Sforza & Edwards, 1967; Saitou & Imanishi, 1989);
- distância de Wagner [DW] (Farris, 1972; Swofford, 1981; Tateno et al., 1982; Nei et al., 1983; Faith, 1985; Sourdis & Krimbas, 1987);
- "neighborliness" [ST] (Sattah & Tversky, 1977; Fitch, 1981);
- "neighbor-joining" [NJ] (Saitou & Nei, 1987);
B) critérios qualitativos: baseados em otimização dos dados;
- máxima parcimônia [MP] (Fitch, 1971);
- parcimônia evolutiva [PE] (Lake, 1988);
- máxima verossimilhança
[MV] (Felsenstein, 1981).
Nesse curso daremos ênfase aos critérios qualitativos, especialmente aos métodos de máxima parcimônia e máxima verossimilhança. Informações mais detalhadas sobre os critérios quantitativos podem ser encontradas no endereço http://adi-38.bio.ib.usp.br/sbg2k/e em algumas das referências bibliograficas citadas ao final desta "apostila".
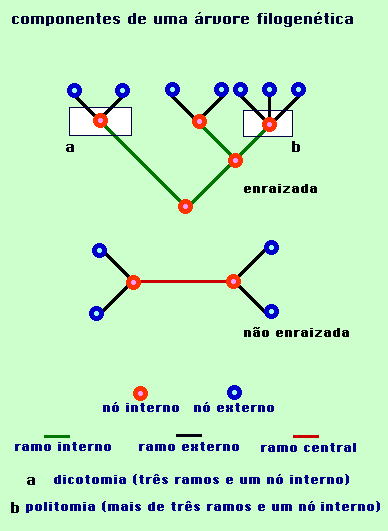
A representação gráfica de reconstrução filogenética geralmente é em forma de uma "árvore" com uma topologia específica, seja ela enraizada ou não. As características gerais de uma árvore filogenética estão apresentadas na seguir: