Figura 1
O princípio da MV
para inferências fiogenéticas é avaliar a probabilidade
de que um determinado modelo de mudanças evolutivas possa explicar
a origem dos dados observados. Estimativas
de MV foram utilizadas pela primeira vez por Cavalli-Sforza & Edwards
em 1967, embora não utilizando dados de seqüência de
macromoléculas. Foi em 1981 que Felsenstein formalizou a utilização
de MV utilizando seqüências de nucleotídeos e em 1990
que Kishino e colaboradores apresentaram pela primeira vez a utilização
de MV para seqüências de aminoácidos.
O objetivo da MV é inferir a história ou o conjunto de histórias evolutivas que sejam os mais consistentes em relação aos dados estudados. Para a aplicação da MV, é necessário que um modelo concreto de mudanças evolutivas que leve à conversão de uma seqüência em outra seja especificado. O modelo pede ser:
a) completamente definido
b) conter uma série de parâmetros os quais serão estimados a partir dos dados.
Sumarizando, na metodologia de MV, os modelos de mudanças evolutivas são avaliados quanto à sua probabilidade de explicar um conjunto de dados de forma que reflita a história evolutiva mais próxima da realidade, ou seja, a história mais verossímil (daí verossimilhança). Nessa avaliação, os modelos recebem valores de verossimilhança e aquele que apresentar o melhor valor é o que será utilizado para se inferir a árvore filogenética.
Como funciona a avaliação da verossimilhança? Vejamos:
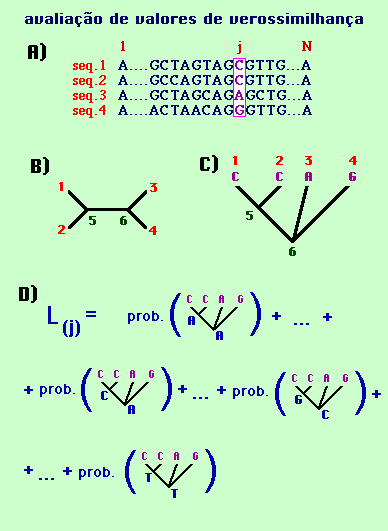
Imagine o conjunto de seqüências de nucleotídeos apresentado na figura 1A. Suponha que queiramos avaliar a verossimilhança da árvore não enraizada apresentada na figura 1B, ou seja, queremos avaliar qual a probabilidade de que essa árvore tenha dado origem às seqüências 1, 2, 3 e 4 com base num modelo evolutivo "X" escolhido. Embora a verossimilhança independa de raiz, por questões práticas convém enraizar a árvore a partir de um dos nós internos (figura 1C).
Figura 1
Se considerarmos a suposição de que todos os sítios evoluem independentemente, podemos calcular o valor de verossimilhança para cada um dos sítios e, ao final, combinar todos os valores de verossimilhança. Por exemplo, tomemos o sítio "j" do exemplo (figura 1A). Todos os cenários evolutivos possíveis devem ser considerados. Claro que uns são mais plausíveis que outros, mas TODOS devem ser considerados, pois todos apresentam alguma probabilidade de ter gerado as seqüências atuais; assim, todas as quatro bases deverão ser consideradas em cada um dos nós internos (figura 1D). Dessa maneira, temos 4 x 4 = 16 possibilidades a considerar.
Como as mudanças ao longo dos ramos são independentes, a probabilidade de um dado cenário evolutivo será igual ao produto das probabilidades de mudanças necessárias (L= likelihood, verossimilhança em inglês):
![]()
Como a probabilidade de cada uma das possíveis situações são extremamente baixas, costuma-se calcular o logaritmo da verossimilhança. Assim, as probabilidades são acumuladas na forma da soma dos logaritmos de cada um dos valores de verossimilhança:
![]()